Думаю, что многие читатели Хабра уже слышали о биоинформатике, возможно даже непосредственно о задаче сборки генома. Множество людей по всем миру занято написанием геномных ассемблеров — программ, интерпретирующих сырые данные машин для секвенирования и выдающих в результате последовательность ДНК изучаемого организма. Однако, в большинстве случаев, геном целиком «из коробки» получить не удается. В этой статье я постараюсь объяснить, почему же геном нельзя собрать одним щелчком мыши и опишу процесс его «финиширования» — пожалуй, самый трудоемкий этап во всей сборке, порой длящийся несколько лет.
Также, я расскажу, как мы иногда можем существенно облегчить этот процесс, используя уже собранные геномы близкородственных организмов. Этой задачей я занимался в рамках написания своей магистерской диссертации в Санкт-Петербургском Академическом Университете, а обучение проходило совместно с Институтом Биоинформатики. Поскольку получившийся алгоритм достаточно специфичен, я начну с описания проблемы в целом, дам обзор некоторых «хардварных» методов ее решения, а затем немного расскажу о том, что же получилось у меня.
Итак, мы хотим собрать геном. На Хабре уже есть пара хороших публикаций на тему геномных ассемблеров: раз, два.
Если биоинформатика для вас нечто новое — крайне рекомендую начать сначала с них, так как в текущей статье подразумевается знание некоторых базовых понятий.
На всякий случай напомню о том, что геномный ассемблер (сборщик) — это программа, которая принимает на вход короткие (несколько сотен нуклеотидов) перекрывающиеся кусочки генома, называемые ридами (reads), и собирает их в единую последовательность. Точнее, пытается собрать — в большинстве случаев даже для относительно небольших бактериальных геномов (несколько миллионов нуклеотидов) цельной последовательности не получается. Это происходит по ряду причин, описанных в статьях выше. Я же кратко скажу, что так бывает из-за наличия в геноме повторяющихся кусков, участков, физически сложных для прочтения, а также ошибок в процессе секвенирования.
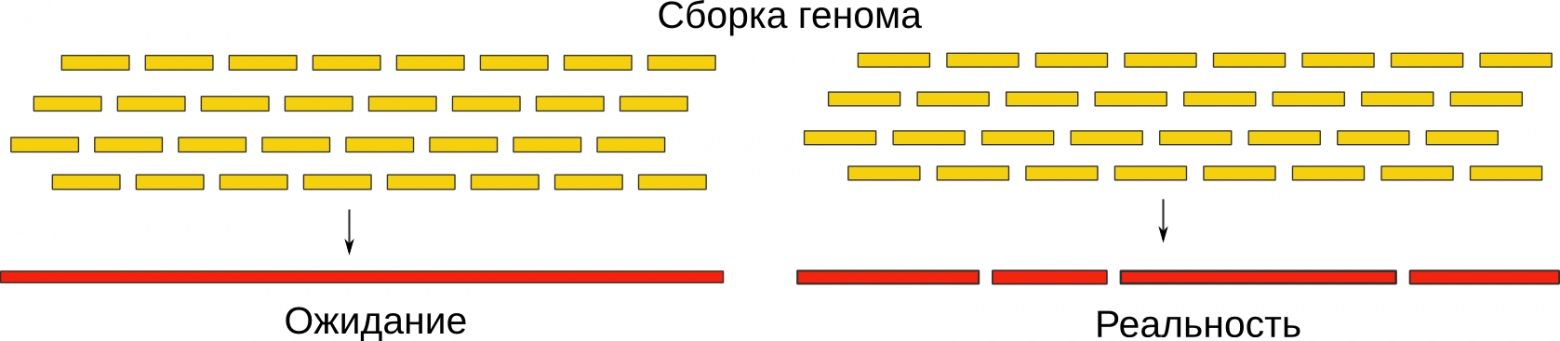
В итоге, вместо того, чтобы выдать нам геном целиком, мы получаем его кусочки, называемые контиги (contigs). Они уже гораздо длиннее (десятки и сотни тысяч нуклеотидов), и в своей совокупности они составляют оригинальную последовательность. Однако, правильный их порядок нам неизвестен. Что же делать дальше?
Для начала зададимся вопросом: а зачем вообще нам цельный геном? Ведь длины контигов уже достаточно, к примеру, для поиска генов внутри них. С другой стороны, некоторые гены все-таки будут повреждены из-за фрагментации. Но, что еще важно, для некоторых исследований необходима именно структура генома, т.е. порядок расположения в нем изучаемых генов.
К примеру, это важно при изучении эволюции. Как мы хорошо знаем, ДНК со временем мутирует. Чаще всего происходят маленькие изменения: один нуклеотид заменяется на другой, или же несколько подряд идущих нуклеотидов вырезаются/вставляются в другое место. Но бывают гораздо более редкие, но серьезные изменения. К примеру, крупный кусок генома может перевернуться или «переехать» на другую позицию. Бывают, что целые хромосомы сливаются друг с другом, или наоборот, распадаются на две части. В своей работе Ханнехалли и Певзнер показали, что геномы человека и мыши разъединяют всего 131 такая перестройка. Согласитесь, не так уж и много.
Теперь, когда я вас заинтересовал, давайте уже соберем этот геном! У нас есть набор контигов — его кусочков, порядок следования которых нам неизвестен. Что будем делать дальше? Нам может помочь технология таргетного секвенирования, позволяющая прочитать определенную и относительно короткую (до нескольких тысяч нуклеотидов) область генома, содержащую определенный паттерн на концах. Для этого используется полимеразная цепная реакция (ПЦР), а затем получившийся продукт прочитывается значительно более дорогим, но надежным методом секвенирования — методом Сэнгера. Запустив такую реакцию и выбрав в качестве паттерна концы двух интересующих нас контигов, мы можем узнать, являются ли они соседними в геноме, а также прочитать недостающую последовательность между ними. Если же наше предположение не верно, и контиги расположены слишком далеко друг от друга — реакция просто-напросто не пойдет.
Но ведь у нас есть сотни (если мы собираем бактерию) и тысячи (млекопитающее) контигов, соответственно десятки тысяч и миллионы возможных их «соединений». На то, чтобы проверить каждое из них, у нас не хватит ни времени, ни денег. Как нам поступить в такой ситуации? Может быть, мы можем как-то оценить, какие соединения вероятнее всего существуют, а какие даже и проверять не стоит?
Разумеется, можем! Существуют разные способы (о некоторых из которых я сейчас и расскажу), которые позволяют объединять контиги в так называемый скаффолд (scaffold), представляющий из себя их упорядоченный набор. Между контигами располагаются «пропуски» (gaps) определенной длинны, символизирующие неизвестную последовательность:
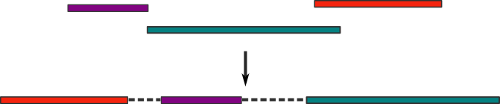
Понятно, что имея такой «черновик» генома, нам будет уже гораздо легче заполнять пропуски в нем. Давайте рассмотрим, какие бывают способы объединения контигов в скаффолды (этот процесс так и называется — скаффолдинг).
Что может быть лучше одного короткого рида? Да разве только два таких же! Давайте разрежем ДНК, которую нам предстоит секвенировать на кусочки некоторой известной нам длины, а затем прочитаем каждый из них с начала и конца. Разумеется, большая часть последовательности между концами останется непрочитанной, зато теперь у нас есть пара ридов, расстояние между которыми нам известно.
Такие пары ридов дают нам новую дополнительную информацию. Давайте «выровняем» (align) их (т.е. найдем вхождение, возможно, неточное) на наши контиги. Если при этом два рида выравниваются на разные контиги, то последние мы можем объединить в единый скаффолд:
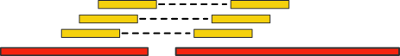
Важным критерием данной технологии является расстояние между парами ридов — так называемая, длина вставки. Чем она больше, тем через более длинные промежутки генома мы можем перескакивать. Однако, с ростом длины вставки существенно возрастает стоимость эксперимента, а также количество ошибок.
В последнее время начинают появляться технологии, позволяющие получать гораздо более длинные риды (до нескольких десятков и даже сотен тысяч пар оснований) в относительно больших количествах. К примеру, такую возможность предоставляет технология PacBio. Однако, во всех них пока что есть два существенных недостатка: во-первых, дороговизна процесса, а во-вторых, наличие большого количества ошибок.
Собирать геном с таким множеством неточностей — та еще задача. Некоторые пошли другим путем — созданием гибридных сборок. В них комбинируются обычные короткие риды, а также длинные, но менее точные. Идея понятна интуитивно: сначала, как обычно, мы выполняем сборку, используя только короткие риды, а затем получившиеся контиги выравниваем на длинные риды. Это, как и в случае с парными ридами, дает нам информацию о расположении контигов друг относительно друга. Также, становится доступна и часть генома между ними:
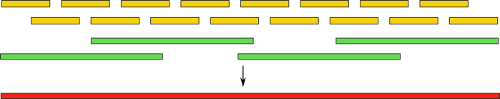
Используя такую технику, не так давно группе ученых удалось полностью автоматически собрать бактериальный геном. Также, есть заметные успехи в сборке с использованием одних только длинных ридов. Однако, вся технология пока, к сожалению, остается уделом избранных из-за своей дороговизны и малой распространенности.
Hi-C — совсем свежая и многообещающая технология, которая позволяет измерить взаимодействие кусков генома в пространстве. Как вы, наверное, знаете, ДНК — не просто длинная линейная молекула, она еще и сложным образом упакована в пространственную структуру, которая и называется хромосомой. Не вдаваясь в подробности, скажу, что Hi-C позволяет получать пары ридов, для которых соответствующие куски генома находятся близко в пространстве, но совсем не обязательно они будут рядом, если мы растянем всю молекулу в одну линию. На картинке ниже показаны интенсивности взаимодействия участков хромосомы друг с другом (по обоим осям — линейные координаты на хромосоме):
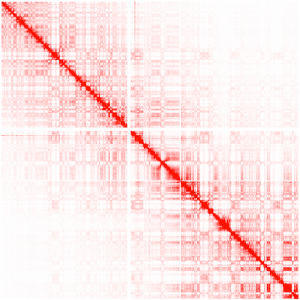
Так мы можем получить довольно интересную информацию, отражающую форму упаковки ДНК в клетке. Как это может быть использовано для скаффолдинга? Да очень просто: куски ДНК, которые линейно близки друг к другу, будут все-таки в среднем чаще взаимодействовать между собой (что мы и видим на картинке выше). Опять же, не буду пускаться в долгие объяснения и отошлю читателя к оригинальный статье на эту тему.
Наконец, мы очень плавно подошли к теме моей работы. Со времени появления первых технологий секвенирования, человечество успело накопить довольно много данных. Уже существует большое количество полностью (или почти полностью) собранных геномов различных организмов. Почему бы не воспользоваться этой информацией, вместо того, чтобы проводить дорогостоящие эксперименты?
И воспользоваться можно! Существует множество чисто вычислительных методов под общим названием «референсная сборка» (reference-assisted assembly), которые подразумевают использование последовательностей уже собранных родственных организмов для улучшения качества сборки нового образца. Базовая идея здесь схожа с той, что используется при сборке с длинными ридами: мы находим выравнивание контигов теперь уже на референсный геном, и объединяем их в соответствующем порядке:
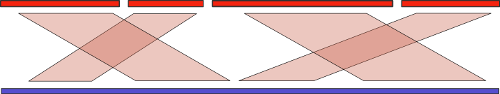
Но, как мы знаем, эволюция не стоит на месте, и различия в собираемом и референсном геноме могут оказаться существенными. Во-первых, это может привести к сложностям при поиске выравнивания. Но это еще полбеды. В самом начала статьи я упоминал про крупномасштабные геномные перестройки. Понятно, что если такие перестройки между собираемым и референсным геномом присутствуют, то мы рискуем получить совершенно неправильный результат. Стало быть, нам нужен алгоритм, который анализирует такие перестройки и старается «расставить все по своим местам».
Написанием такого алгоритма я и занимался в качестве своей магистерской диссертации. Главным его отличием от существующих решений является использование нескольких референсных геномов одновременно. Это дает нам дополнительную информацию о том, как геномы менялись в ходе эволюции, а значит позволяет более достоверно оценить порядок контигов и в нашем геноме.
Не вдаваясь в подробности, я попытаюсь объяснить, почему же несколько референсов лучше, чем один. Начнем с того, что между всеми геномами, которые даны нам на входе, имеется какая-то эволюционная взаимосвязь. Проще говоря, мы можем построить филогенетическое дерево, в листьях которого будут располагаться наши геномы, как референсные — R, так и целевой — T. Дерево показывает их направленную эволюцию от некоторого общего предка, а длинна ветвей соответствует длительности процессов:
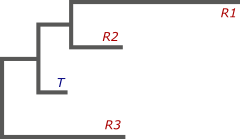
Теперь, зная структуру референсных геномов, нам необходимо восстановить целевой. Чтобы сделать это, необходимо понять, как проходила эволюция всего семейства. Нам важно предсказать, в каких местах филогенетического дерева происходили геномные перестройки, последствия которых мы наблюдаем в листьях. Зная это, мы сможем определить, какие перестройки произошли до, а какие после отделения собираемого нами генома в отдельную ветвь. А значит, сможем и сказать, какие из них вероятнее всего присутствуют в нашей сборке.
По результатам работы была написана статья, которая и стала моей магистерской диссертацией. Саму программу можно найти на гитхабе — fenderglass.github.io/Ragout. Детальное описание алгоритма, к сожалению, выходит за рамки публикации формата Хабра, и было бы не слишком понятно без дополнительного бэкграунда. Но если данная статья вызовет интерес у сообщества, то в дальнейшем я продолжу писать про биоинформатические алгоритмы, связанные с этой темой, и буду постепенно углубляться в область.
P.S. Хотел написать еще и в Биоинформатику, но не хватает кармы. — уже написал
Также, я расскажу, как мы иногда можем существенно облегчить этот процесс, используя уже собранные геномы близкородственных организмов. Этой задачей я занимался в рамках написания своей магистерской диссертации в Санкт-Петербургском Академическом Университете, а обучение проходило совместно с Институтом Биоинформатики. Поскольку получившийся алгоритм достаточно специфичен, я начну с описания проблемы в целом, дам обзор некоторых «хардварных» методов ее решения, а затем немного расскажу о том, что же получилось у меня.
Введение
Итак, мы хотим собрать геном. На Хабре уже есть пара хороших публикаций на тему геномных ассемблеров: раз, два.
Если биоинформатика для вас нечто новое — крайне рекомендую начать сначала с них, так как в текущей статье подразумевается знание некоторых базовых понятий.
На всякий случай напомню о том, что геномный ассемблер (сборщик) — это программа, которая принимает на вход короткие (несколько сотен нуклеотидов) перекрывающиеся кусочки генома, называемые ридами (reads), и собирает их в единую последовательность. Точнее, пытается собрать — в большинстве случаев даже для относительно небольших бактериальных геномов (несколько миллионов нуклеотидов) цельной последовательности не получается. Это происходит по ряду причин, описанных в статьях выше. Я же кратко скажу, что так бывает из-за наличия в геноме повторяющихся кусков, участков, физически сложных для прочтения, а также ошибок в процессе секвенирования.

В итоге, вместо того, чтобы выдать нам геном целиком, мы получаем его кусочки, называемые контиги (contigs). Они уже гораздо длиннее (десятки и сотни тысяч нуклеотидов), и в своей совокупности они составляют оригинальную последовательность. Однако, правильный их порядок нам неизвестен. Что же делать дальше?
Для начала зададимся вопросом: а зачем вообще нам цельный геном? Ведь длины контигов уже достаточно, к примеру, для поиска генов внутри них. С другой стороны, некоторые гены все-таки будут повреждены из-за фрагментации. Но, что еще важно, для некоторых исследований необходима именно структура генома, т.е. порядок расположения в нем изучаемых генов.
К примеру, это важно при изучении эволюции. Как мы хорошо знаем, ДНК со временем мутирует. Чаще всего происходят маленькие изменения: один нуклеотид заменяется на другой, или же несколько подряд идущих нуклеотидов вырезаются/вставляются в другое место. Но бывают гораздо более редкие, но серьезные изменения. К примеру, крупный кусок генома может перевернуться или «переехать» на другую позицию. Бывают, что целые хромосомы сливаются друг с другом, или наоборот, распадаются на две части. В своей работе Ханнехалли и Певзнер показали, что геномы человека и мыши разъединяют всего 131 такая перестройка. Согласитесь, не так уж и много.
Черновик генома
Теперь, когда я вас заинтересовал, давайте уже соберем этот геном! У нас есть набор контигов — его кусочков, порядок следования которых нам неизвестен. Что будем делать дальше? Нам может помочь технология таргетного секвенирования, позволяющая прочитать определенную и относительно короткую (до нескольких тысяч нуклеотидов) область генома, содержащую определенный паттерн на концах. Для этого используется полимеразная цепная реакция (ПЦР), а затем получившийся продукт прочитывается значительно более дорогим, но надежным методом секвенирования — методом Сэнгера. Запустив такую реакцию и выбрав в качестве паттерна концы двух интересующих нас контигов, мы можем узнать, являются ли они соседними в геноме, а также прочитать недостающую последовательность между ними. Если же наше предположение не верно, и контиги расположены слишком далеко друг от друга — реакция просто-напросто не пойдет.
Но ведь у нас есть сотни (если мы собираем бактерию) и тысячи (млекопитающее) контигов, соответственно десятки тысяч и миллионы возможных их «соединений». На то, чтобы проверить каждое из них, у нас не хватит ни времени, ни денег. Как нам поступить в такой ситуации? Может быть, мы можем как-то оценить, какие соединения вероятнее всего существуют, а какие даже и проверять не стоит?
Разумеется, можем! Существуют разные способы (о некоторых из которых я сейчас и расскажу), которые позволяют объединять контиги в так называемый скаффолд (scaffold), представляющий из себя их упорядоченный набор. Между контигами располагаются «пропуски» (gaps) определенной длинны, символизирующие неизвестную последовательность:

Понятно, что имея такой «черновик» генома, нам будет уже гораздо легче заполнять пропуски в нем. Давайте рассмотрим, какие бывают способы объединения контигов в скаффолды (этот процесс так и называется — скаффолдинг).
Технологии скаффолдинга
Библиотеки парных ридов
Что может быть лучше одного короткого рида? Да разве только два таких же! Давайте разрежем ДНК, которую нам предстоит секвенировать на кусочки некоторой известной нам длины, а затем прочитаем каждый из них с начала и конца. Разумеется, большая часть последовательности между концами останется непрочитанной, зато теперь у нас есть пара ридов, расстояние между которыми нам известно.
Такие пары ридов дают нам новую дополнительную информацию. Давайте «выровняем» (align) их (т.е. найдем вхождение, возможно, неточное) на наши контиги. Если при этом два рида выравниваются на разные контиги, то последние мы можем объединить в единый скаффолд:
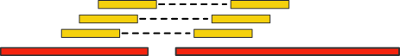
Важным критерием данной технологии является расстояние между парами ридов — так называемая, длина вставки. Чем она больше, тем через более длинные промежутки генома мы можем перескакивать. Однако, с ростом длины вставки существенно возрастает стоимость эксперимента, а также количество ошибок.
Длинные риды
В последнее время начинают появляться технологии, позволяющие получать гораздо более длинные риды (до нескольких десятков и даже сотен тысяч пар оснований) в относительно больших количествах. К примеру, такую возможность предоставляет технология PacBio. Однако, во всех них пока что есть два существенных недостатка: во-первых, дороговизна процесса, а во-вторых, наличие большого количества ошибок.
Собирать геном с таким множеством неточностей — та еще задача. Некоторые пошли другим путем — созданием гибридных сборок. В них комбинируются обычные короткие риды, а также длинные, но менее точные. Идея понятна интуитивно: сначала, как обычно, мы выполняем сборку, используя только короткие риды, а затем получившиеся контиги выравниваем на длинные риды. Это, как и в случае с парными ридами, дает нам информацию о расположении контигов друг относительно друга. Также, становится доступна и часть генома между ними:
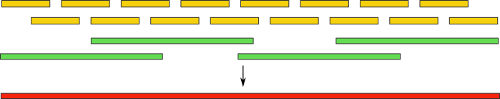
Используя такую технику, не так давно группе ученых удалось полностью автоматически собрать бактериальный геном. Также, есть заметные успехи в сборке с использованием одних только длинных ридов. Однако, вся технология пока, к сожалению, остается уделом избранных из-за своей дороговизны и малой распространенности.
Hi-C
Hi-C — совсем свежая и многообещающая технология, которая позволяет измерить взаимодействие кусков генома в пространстве. Как вы, наверное, знаете, ДНК — не просто длинная линейная молекула, она еще и сложным образом упакована в пространственную структуру, которая и называется хромосомой. Не вдаваясь в подробности, скажу, что Hi-C позволяет получать пары ридов, для которых соответствующие куски генома находятся близко в пространстве, но совсем не обязательно они будут рядом, если мы растянем всю молекулу в одну линию. На картинке ниже показаны интенсивности взаимодействия участков хромосомы друг с другом (по обоим осям — линейные координаты на хромосоме):

Так мы можем получить довольно интересную информацию, отражающую форму упаковки ДНК в клетке. Как это может быть использовано для скаффолдинга? Да очень просто: куски ДНК, которые линейно близки друг к другу, будут все-таки в среднем чаще взаимодействовать между собой (что мы и видим на картинке выше). Опять же, не буду пускаться в долгие объяснения и отошлю читателя к оригинальный статье на эту тему.
Референсная сборка
Наконец, мы очень плавно подошли к теме моей работы. Со времени появления первых технологий секвенирования, человечество успело накопить довольно много данных. Уже существует большое количество полностью (или почти полностью) собранных геномов различных организмов. Почему бы не воспользоваться этой информацией, вместо того, чтобы проводить дорогостоящие эксперименты?
И воспользоваться можно! Существует множество чисто вычислительных методов под общим названием «референсная сборка» (reference-assisted assembly), которые подразумевают использование последовательностей уже собранных родственных организмов для улучшения качества сборки нового образца. Базовая идея здесь схожа с той, что используется при сборке с длинными ридами: мы находим выравнивание контигов теперь уже на референсный геном, и объединяем их в соответствующем порядке:
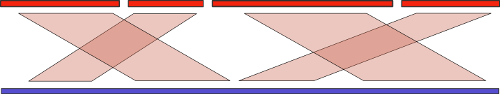
Но, как мы знаем, эволюция не стоит на месте, и различия в собираемом и референсном геноме могут оказаться существенными. Во-первых, это может привести к сложностям при поиске выравнивания. Но это еще полбеды. В самом начала статьи я упоминал про крупномасштабные геномные перестройки. Понятно, что если такие перестройки между собираемым и референсным геномом присутствуют, то мы рискуем получить совершенно неправильный результат. Стало быть, нам нужен алгоритм, который анализирует такие перестройки и старается «расставить все по своим местам».
Написанием такого алгоритма я и занимался в качестве своей магистерской диссертации. Главным его отличием от существующих решений является использование нескольких референсных геномов одновременно. Это дает нам дополнительную информацию о том, как геномы менялись в ходе эволюции, а значит позволяет более достоверно оценить порядок контигов и в нашем геноме.
Не вдаваясь в подробности, я попытаюсь объяснить, почему же несколько референсов лучше, чем один. Начнем с того, что между всеми геномами, которые даны нам на входе, имеется какая-то эволюционная взаимосвязь. Проще говоря, мы можем построить филогенетическое дерево, в листьях которого будут располагаться наши геномы, как референсные — R, так и целевой — T. Дерево показывает их направленную эволюцию от некоторого общего предка, а длинна ветвей соответствует длительности процессов:

Теперь, зная структуру референсных геномов, нам необходимо восстановить целевой. Чтобы сделать это, необходимо понять, как проходила эволюция всего семейства. Нам важно предсказать, в каких местах филогенетического дерева происходили геномные перестройки, последствия которых мы наблюдаем в листьях. Зная это, мы сможем определить, какие перестройки произошли до, а какие после отделения собираемого нами генома в отдельную ветвь. А значит, сможем и сказать, какие из них вероятнее всего присутствуют в нашей сборке.
По результатам работы была написана статья, которая и стала моей магистерской диссертацией. Саму программу можно найти на гитхабе — fenderglass.github.io/Ragout. Детальное описание алгоритма, к сожалению, выходит за рамки публикации формата Хабра, и было бы не слишком понятно без дополнительного бэкграунда. Но если данная статья вызовет интерес у сообщества, то в дальнейшем я продолжу писать про биоинформатические алгоритмы, связанные с этой темой, и буду постепенно углубляться в область.
