Если спросить случайного прохожего, что такое биология, он наверняка ответит что-то вроде «наука о живой природе». Про информатику скажет, что она имеет дело с компьютерами и информацией. Если мы не побоимся быть навязчивыми и зададим ему третий вопрос – что такое биоинформатика? – тут-то он наверняка и растеряется. Логично: про эту область знаний даже в ЕРАМ знает далеко не каждый – хотя в нашей компании и биоинформатики есть. Давайте разбираться, для чего эта наука нужна человечеству вообще и ЕРАМ в частности: в конце концов, вдруг нас на улице об этом спросят.
Чтобы провести исследование, биологам уже недостаточно взять анализы и посмотреть в микроскоп. Современная биология имеет дело с колоссальными объемами данных. Часто обработать их вручную просто невозможно, поэтому многие биологические задачи решаются вычислительными методами. Не будем далеко ходить: молекула ДНК настолько мала, что разглядеть ее под световым микроскопом нельзя. А если и можно (под электронным), всё равно визуальное изучение не помогает решить многих задач.
ДНК человека состоит из трех миллиардов нуклеотидов – чтобы вручную проанализировать их все и найти нужный участок, не хватит и целой жизни. Ну, может и хватит – одной жизни на анализ одной молекулы – но это слишком долго, дорого и малопродуктивно, так что геном анализируют при помощи компьютеров и вычислений.
Биоинформатика — это и есть весь набор компьютерных методов для анализа биологических данных: прочитанных структур ДНК и белков, микрофотографий, сигналов, баз данных с результатами экспериментов и т. д.

Иногда секвенировать ДНК нужно, чтобы подобрать правильное лечение. Одно и то же заболевание, вызванное разными наследственными нарушениями или воздействием среды, нужно лечить по-разному. А еще в геноме есть участки, которые не связаны с развитием болезни, но, например, отвечают за реакцию на определенные виды терапии и лекарств. Поэтому разные люди с одним и тем же заболеванием могут по-разному реагировать на одинаковое лечение.
Еще биоинформатика нужна, чтобы разрабатывать новые лекарства. Их молекулы должны иметь определенную структуру и связываться с определенным белком или участком ДНК. Смоделировать структуру такой молекулы помогают вычислительные методы.
Достижения биоинформатики широко применяют в медицине, в первую очередь в терапии рака. В ДНК зашифрована информация о предрасположенности и к другим заболеваниям, но над лечением рака работают больше всего. Это направление считается самым перспективным, финансово привлекательным, важным – и самым сложным.
В ЕРАМ биоинформатикой занимается подразделение Life Sciences. Там разрабатывают программное обеспечение для фармкомпаний, биологических и биотехнологических лабораторий всех масштабов — от стартапов до ведущих мировых компаний. Справиться с такой задачей могут только люди, которые разбираются в биологии, умеют составлять алгоритмы и программировать.
Биоинформатики – гибридные специалисты. Сложно сказать, какое знание для них первично: биология или информатика. Если так ставить вопрос, им нужно знать и то и другое. В первую очередь важны, пожалуй, аналитический склад ума и готовность много учиться. В ЕРАМ есть и биологи, которые доучились информатике, и программисты с математиками, которые дополнительно изучали биологию.
Мария Зуева, разработчик:
«Я получила стандартное ИТ-образование, потом училась на курсах ЕРАМ Java Lab, где увлеклась машинным обучением и Data Science. Когда я выпускалась из лаборатории, мне сказали: «Сходи в Life Sciences, там занимаются биоинформатикой и как раз набирают людей». Не лукавлю: тогда я услышала слово «биоинформатика» в первый раз. Прочитала про нее на Википедии и пошла.
Тогда в подразделение набрали целую группу новичков, и мы вместе изучали биоинформатику. Начали с повторения школьной программы про ДНК и РНК, затем подробно разбирали существующие в биоинформатике задачи, подходы к их решению и алгоритмы, учились работать со специализированным софтом».
Геннадий Захаров, бизнес-аналитик:
«По образованию я биофизик, в 2012-м защитил кандидатскую по генетике. Какое-то время работал в науке, занимался исследованиями – и продолжаю до сих пор. Когда появилась возможность применить научные знания в производстве, я тут же за нее ухватился.
Для бизнес-аналитика у меня весьма специфическая работа. Например, финансовые вопросы проходят мимо меня, я скорее эксперт по предметной области. Я должен понять, чего от нас хотят заказчики, разобраться в проблеме и составить высокоуровневую документацию – задание для программистов, иногда сделать работающий прототип программы. По ходу проекта я поддерживаю контакт с разработчиками и заказчиками, чтобы те и другие были уверены: команда делает то, что от нее требуется. Фактически я переводчик с языка заказчиков – биологов и биоинформатиков – на язык разработчиков и обратно».
Чтобы понять суть биоинформатических проектов ЕРАМ, сначала нужно разобраться, как секвенируют геном. Дело в том, что проекты, о которых мы будем говорить, напрямую связаны с чтением генома. Обратимся за объяснением к биоинформатикам.
Михаил Альперович, глава юнита биоинформатики:
«Представьте, что у вас есть десять тысяч экземпляров «Войны и мира». Вы пропустили их через шредер, хорошенько перемешали, наугад вытащили из этой кучи ворох бумажных полосок и пытаетесь собрать из них исходный текст. Вдобавок у вас есть рукопись «Войны и мира». Текст, который вы соберете, нужно будет сравнить с ней, чтобы отловить опечатки (а они обязательно будут). Примерно так же читают ДНК современные машины-секвенаторы. ДНК выделяют из клеточных ядер и делят на фрагменты по 300–500 пар нуклеотидов (мы помним, что в ДНК нуклеотиды связаны друг с другом попарно). Молекулы дробят, потому что ни одна современная машина не может прочитать геном от начала до конца. Последовательность слишком длинная, и по мере ее прочтения накапливаются ошибки.
Вспоминаем «Войну и мир» после шредера. Чтобы восстановить исходный текст романа, нам нужно прочитать и расположить в правильном порядке все кусочки романа. Получается, что мы читаем книгу несколько раз по крошечным фрагментам. То же с ДНК: каждый участок последовательности секвенатор прочитывает с многократным перекрытием – ведь мы анализируем не одну, а множество молекул ДНК.
Полученные фрагменты выравнивают – «прикладывают» каждый из них к эталонному геному и пытаются понять, какому участку эталона соответствует прочитанный фрагмент. Затем в выравненных фрагментах находят вариации – значащие отличия прочтений от эталонного генома (опечатки в книге по сравнению с эталонной рукописью). Этим занимаются программы – вариант-коллеры (от англ. variant caller – выявитель мутаций). Это самая сложная часть анализа, поэтому различных программ – вариант-коллеров много и их постоянно совершенствуют и разрабатывают новые.
Подавляющее большинство найденных мутаций нейтральны и ни на что не влияют. Но есть и такие, в которых зашифрованы предрасположенность к наследственным заболеваниям или способность откликаться на разные виды терапии».

Для анализа берут образец, в котором находится много клеток — а значит, и копий полного набора ДНК клетки. Каждый маленький фрагмент ДНК прочитывают несколько раз, чтобы минимизировать вероятность ошибки. Если пропустить хотя бы одну значащую мутацию, можно поставить пациенту неверный диагноз или назначить неподходящее лечение. Прочитать каждый фрагмент ДНК по одному разу слишком мало: единственное прочтение может быть неправильным, и мы об этом не узнаем. Если мы прочитаем тот же фрагмент дважды и получим один верный и один неверный результат, нам будет сложно понять, какое из прочтений правдивое. А если у нас сто прочтений и в 95 из них мы видим один и тот же результат, мы понимаем, что он и есть верный.
Геннадий Захаров:
«Для анализа раковых заболеваний секвенировать нужно и здоровую, и больную клетку. Рак появляется в результате мутаций, которые клетка накапливает в течение своей жизни. Если в клетке испортились механизмы, отвечающие за ее рост и деление, то клетка начинает неограниченно делиться вне зависимости от потребностей организма, т. е. становится раковой опухолью. Чтобы понять, чем именно вызван рак, у пациента берут образец здоровой ткани и раковой опухоли. Оба образца секвенируют, сопоставляют результаты и находят, чем один отличается от другого: какой молекулярный механизм сломался в раковой клетке. Исходя из этого подбирают лекарство, которое эффективно против клеток с “поломкой”».
У подразделения биоинформатики в ЕРАМ есть и производственные, и опенсорс-проекты. Причем часть производственного проекта может перерасти в опенсорс, а опенсорсный проект – стать частью производства (например, когда продукт ЕРАМ с открытым кодом нужно интегрировать в инфраструктуру клиента).
Для одного из клиентов – крупной фармацевтической компании – ЕРАМ модернизировал программу вариант-коллер. Ее особенность в том, что она способна находить мутации, недоступные другим аналогичным программам. Изначально программа была написана на языке Perl и обладала сложной логикой. В ЕРАМ программу переписали на Java и оптимизировали – теперь она работает в 20, если не в 30 раз быстрее.
Исходный код программы доступен на GitHub.
Для визуализации структуры молекул в 3D есть много десктоп- и веб-приложений. Представлять, как молекула выглядит в пространстве, крайне важно, например, для разработки лекарств. Предположим, нам нужно синтезировать лекарство, обладающее направленным действием. Сначала нам потребуется спроектировать молекулу этого лекарства и убедиться, что она будет взаимодействовать с нужными белками именно так, как нужно. В жизни молекулы трехмерные, поэтому анализируют их тоже в виде трехмерных структур.
Для 3D-просмотра молекул ЕРАМ сделал онлайн-инструмент, который изначально работал только в окне браузера. Потом на основании этого инструмента разработали версию, которая позволяет визуализировать молекулы в очках виртуальной реальности HTC Vive. К очкам прилагаются контроллеры, которыми молекулу можно поворачивать, перемещать, подставлять к другой молекуле, поворачивать отдельные части молекулы. Делать всё это в 3D куда удобнее, чем на плоском мониторе. Эту часть проекта биоинформатики ЕРАМ делали совместно с подразделением Virtual Reality, Augmented Reality and Game Experience Delivery.
Программа только готовится к публикации на GitHub, зато пока есть ссылка, по которой можно посмотреть ее демо-версию.
Как выглядит работа с приложением, можно узнать из видео.
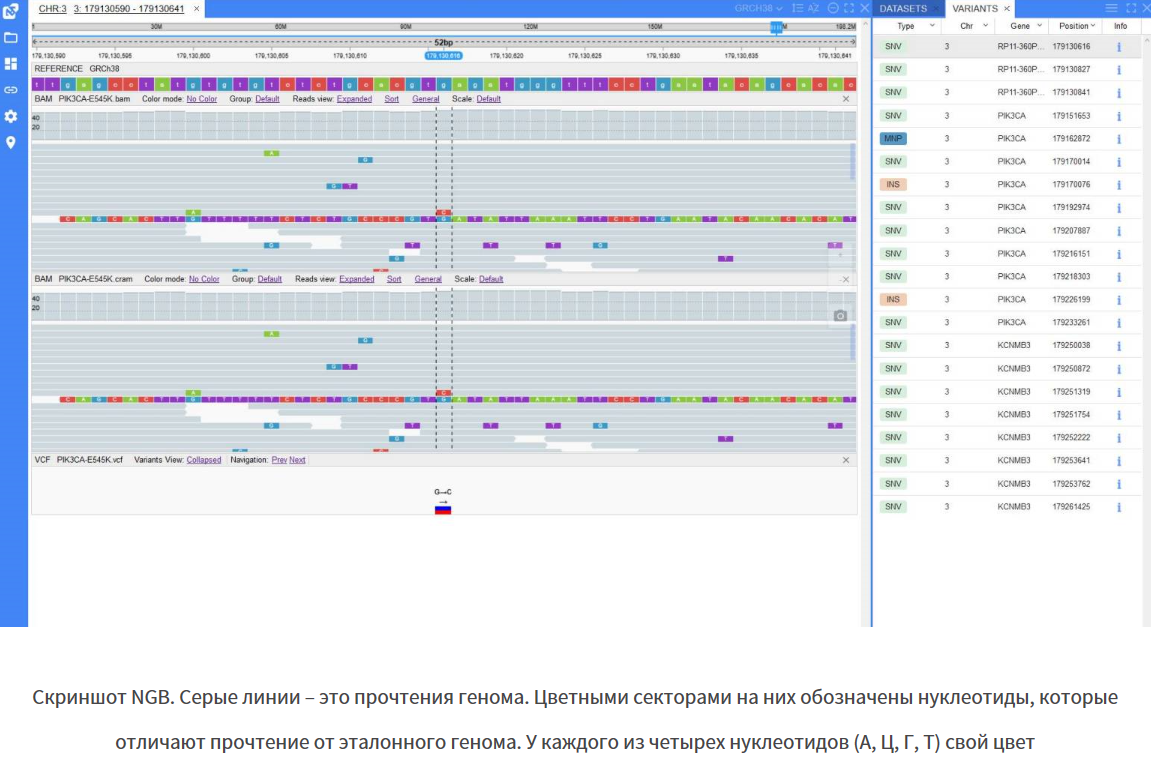
Геномный браузер визуализирует отдельные прочтения ДНК, вариации и другую информацию, сгенерированную утилитами для анализа генома. Когда прочтения сопоставлены с эталонным геномом и мутации найдены, ученому остается проконтролировать, правильно ли сработали машины и алгоритмы. От того, насколько точно выявлены мутации в геноме, зависит, какой диагноз поставят пациенту или какое лечение ему назначат. Поэтому в клинической диагностике контролировать работу машин должен ученый, а помогает ему в этом геномный браузер.
Биоинформатикам-разработчикам геномный браузер помогает анализировать сложные случаи, чтобы найти ошибки в работе алгоритмов и понять, как их можно улучшить.
Новый геномный браузер NGB (New Genome Browser) от ЕРАМ работает в вебе, но по скорости и функционалу не уступает десктопным аналогам. Это продукт, которого не хватало на рынке: предыдущие онлайновые инструменты работали медленнее и умели делать меньше, чем десктопные. Сейчас многие клиенты выбирают веб-приложения из соображений безопасности. Онлайн-инструмент позволяет ничего не устанавливать на рабочий компьютер ученого. С ним можно работать из любой точки мира, зайдя на корпоративный портал. Ученому не обязательно всюду возить за собой рабочий компьютер и скачивать на него все необходимые данные, которых может быть очень много.
Геннадий Захаров, бизнес-аналитик:
«Над опенсорсными утилитами я работал частично как заказчик: ставил задачу. Я изучал лучшие решения на рынке, анализировал их преимущества и недостатки, искал, как можно их усовершенствовать. Нам нужно было сделать веб-решения не хуже десктопных аналогов и при этом добавить в них что-то уникальное.
В 3D-просмотрщике молекул это была работа с виртуальной реальностью, а в геномном браузере – улучшенная работа с вариациями. Мутации бывают сложными. Перестройки в раковых клетках иногда затрагивают огромные области. В них появляются лишние хромосомы, куски хромосом и целые хромосомы исчезают или объединяются в случайном порядке. Отдельные куски генома могут копироваться по 10–20 раз. Такие данные, во-первых, сложнее получить из прочтений, а во-вторых, сложнее визуализировать.
Мы разработали визуализатор, который правильно читает информацию о таких протяженных структурных перестройках. Еще мы сделали набор визуализаций, который при контакте хромосом показывает, образовались ли из-за этого контакта гибридные белки. Если протяженная вариация затрагивает несколько белков, мы по клику можем рассчитать и показать, что происходит в результате такой вариации, какие гибридные белки получаются. В других визуализаторах ученым приходилось отслеживать эту информацию вручную, а в NGB – в один клик».
Мы уже говорили, что биоинформатики – гибридные специалисты, которые должны знать и биологию, и информатику. Самообразование играет в этом не последнюю роль. Конечно, в ЕРАМ есть вводный курс в биоинформатику, но рассчитан он на сотрудников, которым эти знания пригодятся на проекте. Занятия проводятся только в Санкт-Петербурге. И всё же, если биоинформатика вам интересна, возможность учиться есть:
1) Вводный курс в генетическую диагностику от компании 23andme.
2) Несколько курсов на Coursera (в том числе пара курсов на русском: введение в биоинформатику и в метагеномику).
3) Курсы на Stepik от института биоинформатики: молекулярная биология и генетика, молекулярная филогенетика, генная инженерия и введение в технологии высокоэффективного секвенирования. Полный список курсов от института можно посмотреть на его официальном сайте.
4) Лекции Павла Певзнера – профессора Калифорнийского университета в Сан-Диего, специалиста в области биоинформатики.
5) Если вы живете в Санкт-Петербурге, можно прийти на гостевые лекции в институт биоинформатики – это бесплатно.
Почему биология перестала справляться без информатики и при чем тут рак
Чтобы провести исследование, биологам уже недостаточно взять анализы и посмотреть в микроскоп. Современная биология имеет дело с колоссальными объемами данных. Часто обработать их вручную просто невозможно, поэтому многие биологические задачи решаются вычислительными методами. Не будем далеко ходить: молекула ДНК настолько мала, что разглядеть ее под световым микроскопом нельзя. А если и можно (под электронным), всё равно визуальное изучение не помогает решить многих задач.
ДНК человека состоит из трех миллиардов нуклеотидов – чтобы вручную проанализировать их все и найти нужный участок, не хватит и целой жизни. Ну, может и хватит – одной жизни на анализ одной молекулы – но это слишком долго, дорого и малопродуктивно, так что геном анализируют при помощи компьютеров и вычислений.
Биоинформатика — это и есть весь набор компьютерных методов для анализа биологических данных: прочитанных структур ДНК и белков, микрофотографий, сигналов, баз данных с результатами экспериментов и т. д.

Иногда секвенировать ДНК нужно, чтобы подобрать правильное лечение. Одно и то же заболевание, вызванное разными наследственными нарушениями или воздействием среды, нужно лечить по-разному. А еще в геноме есть участки, которые не связаны с развитием болезни, но, например, отвечают за реакцию на определенные виды терапии и лекарств. Поэтому разные люди с одним и тем же заболеванием могут по-разному реагировать на одинаковое лечение.
Еще биоинформатика нужна, чтобы разрабатывать новые лекарства. Их молекулы должны иметь определенную структуру и связываться с определенным белком или участком ДНК. Смоделировать структуру такой молекулы помогают вычислительные методы.
Достижения биоинформатики широко применяют в медицине, в первую очередь в терапии рака. В ДНК зашифрована информация о предрасположенности и к другим заболеваниям, но над лечением рака работают больше всего. Это направление считается самым перспективным, финансово привлекательным, важным – и самым сложным.
Биоинформатика в ЕРАМ
В ЕРАМ биоинформатикой занимается подразделение Life Sciences. Там разрабатывают программное обеспечение для фармкомпаний, биологических и биотехнологических лабораторий всех масштабов — от стартапов до ведущих мировых компаний. Справиться с такой задачей могут только люди, которые разбираются в биологии, умеют составлять алгоритмы и программировать.
Биоинформатики – гибридные специалисты. Сложно сказать, какое знание для них первично: биология или информатика. Если так ставить вопрос, им нужно знать и то и другое. В первую очередь важны, пожалуй, аналитический склад ума и готовность много учиться. В ЕРАМ есть и биологи, которые доучились информатике, и программисты с математиками, которые дополнительно изучали биологию.
Как становятся биоинформатиками
Мария Зуева, разработчик:
«Я получила стандартное ИТ-образование, потом училась на курсах ЕРАМ Java Lab, где увлеклась машинным обучением и Data Science. Когда я выпускалась из лаборатории, мне сказали: «Сходи в Life Sciences, там занимаются биоинформатикой и как раз набирают людей». Не лукавлю: тогда я услышала слово «биоинформатика» в первый раз. Прочитала про нее на Википедии и пошла.
Тогда в подразделение набрали целую группу новичков, и мы вместе изучали биоинформатику. Начали с повторения школьной программы про ДНК и РНК, затем подробно разбирали существующие в биоинформатике задачи, подходы к их решению и алгоритмы, учились работать со специализированным софтом».
Геннадий Захаров, бизнес-аналитик:
«По образованию я биофизик, в 2012-м защитил кандидатскую по генетике. Какое-то время работал в науке, занимался исследованиями – и продолжаю до сих пор. Когда появилась возможность применить научные знания в производстве, я тут же за нее ухватился.
Для бизнес-аналитика у меня весьма специфическая работа. Например, финансовые вопросы проходят мимо меня, я скорее эксперт по предметной области. Я должен понять, чего от нас хотят заказчики, разобраться в проблеме и составить высокоуровневую документацию – задание для программистов, иногда сделать работающий прототип программы. По ходу проекта я поддерживаю контакт с разработчиками и заказчиками, чтобы те и другие были уверены: команда делает то, что от нее требуется. Фактически я переводчик с языка заказчиков – биологов и биоинформатиков – на язык разработчиков и обратно».
Как читают геном
Чтобы понять суть биоинформатических проектов ЕРАМ, сначала нужно разобраться, как секвенируют геном. Дело в том, что проекты, о которых мы будем говорить, напрямую связаны с чтением генома. Обратимся за объяснением к биоинформатикам.
Михаил Альперович, глава юнита биоинформатики:
«Представьте, что у вас есть десять тысяч экземпляров «Войны и мира». Вы пропустили их через шредер, хорошенько перемешали, наугад вытащили из этой кучи ворох бумажных полосок и пытаетесь собрать из них исходный текст. Вдобавок у вас есть рукопись «Войны и мира». Текст, который вы соберете, нужно будет сравнить с ней, чтобы отловить опечатки (а они обязательно будут). Примерно так же читают ДНК современные машины-секвенаторы. ДНК выделяют из клеточных ядер и делят на фрагменты по 300–500 пар нуклеотидов (мы помним, что в ДНК нуклеотиды связаны друг с другом попарно). Молекулы дробят, потому что ни одна современная машина не может прочитать геном от начала до конца. Последовательность слишком длинная, и по мере ее прочтения накапливаются ошибки.
Вспоминаем «Войну и мир» после шредера. Чтобы восстановить исходный текст романа, нам нужно прочитать и расположить в правильном порядке все кусочки романа. Получается, что мы читаем книгу несколько раз по крошечным фрагментам. То же с ДНК: каждый участок последовательности секвенатор прочитывает с многократным перекрытием – ведь мы анализируем не одну, а множество молекул ДНК.
Полученные фрагменты выравнивают – «прикладывают» каждый из них к эталонному геному и пытаются понять, какому участку эталона соответствует прочитанный фрагмент. Затем в выравненных фрагментах находят вариации – значащие отличия прочтений от эталонного генома (опечатки в книге по сравнению с эталонной рукописью). Этим занимаются программы – вариант-коллеры (от англ. variant caller – выявитель мутаций). Это самая сложная часть анализа, поэтому различных программ – вариант-коллеров много и их постоянно совершенствуют и разрабатывают новые.
Подавляющее большинство найденных мутаций нейтральны и ни на что не влияют. Но есть и такие, в которых зашифрованы предрасположенность к наследственным заболеваниям или способность откликаться на разные виды терапии».

Для анализа берут образец, в котором находится много клеток — а значит, и копий полного набора ДНК клетки. Каждый маленький фрагмент ДНК прочитывают несколько раз, чтобы минимизировать вероятность ошибки. Если пропустить хотя бы одну значащую мутацию, можно поставить пациенту неверный диагноз или назначить неподходящее лечение. Прочитать каждый фрагмент ДНК по одному разу слишком мало: единственное прочтение может быть неправильным, и мы об этом не узнаем. Если мы прочитаем тот же фрагмент дважды и получим один верный и один неверный результат, нам будет сложно понять, какое из прочтений правдивое. А если у нас сто прочтений и в 95 из них мы видим один и тот же результат, мы понимаем, что он и есть верный.
Геннадий Захаров:
«Для анализа раковых заболеваний секвенировать нужно и здоровую, и больную клетку. Рак появляется в результате мутаций, которые клетка накапливает в течение своей жизни. Если в клетке испортились механизмы, отвечающие за ее рост и деление, то клетка начинает неограниченно делиться вне зависимости от потребностей организма, т. е. становится раковой опухолью. Чтобы понять, чем именно вызван рак, у пациента берут образец здоровой ткани и раковой опухоли. Оба образца секвенируют, сопоставляют результаты и находят, чем один отличается от другого: какой молекулярный механизм сломался в раковой клетке. Исходя из этого подбирают лекарство, которое эффективно против клеток с “поломкой”».
Биоинформатика: производство и опенсорс
У подразделения биоинформатики в ЕРАМ есть и производственные, и опенсорс-проекты. Причем часть производственного проекта может перерасти в опенсорс, а опенсорсный проект – стать частью производства (например, когда продукт ЕРАМ с открытым кодом нужно интегрировать в инфраструктуру клиента).
Проект №1: вариант-коллер
Для одного из клиентов – крупной фармацевтической компании – ЕРАМ модернизировал программу вариант-коллер. Ее особенность в том, что она способна находить мутации, недоступные другим аналогичным программам. Изначально программа была написана на языке Perl и обладала сложной логикой. В ЕРАМ программу переписали на Java и оптимизировали – теперь она работает в 20, если не в 30 раз быстрее.
Исходный код программы доступен на GitHub.
Проект №2: 3D-просмотрщик молекул
Для визуализации структуры молекул в 3D есть много десктоп- и веб-приложений. Представлять, как молекула выглядит в пространстве, крайне важно, например, для разработки лекарств. Предположим, нам нужно синтезировать лекарство, обладающее направленным действием. Сначала нам потребуется спроектировать молекулу этого лекарства и убедиться, что она будет взаимодействовать с нужными белками именно так, как нужно. В жизни молекулы трехмерные, поэтому анализируют их тоже в виде трехмерных структур.
Для 3D-просмотра молекул ЕРАМ сделал онлайн-инструмент, который изначально работал только в окне браузера. Потом на основании этого инструмента разработали версию, которая позволяет визуализировать молекулы в очках виртуальной реальности HTC Vive. К очкам прилагаются контроллеры, которыми молекулу можно поворачивать, перемещать, подставлять к другой молекуле, поворачивать отдельные части молекулы. Делать всё это в 3D куда удобнее, чем на плоском мониторе. Эту часть проекта биоинформатики ЕРАМ делали совместно с подразделением Virtual Reality, Augmented Reality and Game Experience Delivery.
Программа только готовится к публикации на GitHub, зато пока есть ссылка, по которой можно посмотреть ее демо-версию.
Как выглядит работа с приложением, можно узнать из видео.
Проект №3: геномный браузер NGB
Геномный браузер визуализирует отдельные прочтения ДНК, вариации и другую информацию, сгенерированную утилитами для анализа генома. Когда прочтения сопоставлены с эталонным геномом и мутации найдены, ученому остается проконтролировать, правильно ли сработали машины и алгоритмы. От того, насколько точно выявлены мутации в геноме, зависит, какой диагноз поставят пациенту или какое лечение ему назначат. Поэтому в клинической диагностике контролировать работу машин должен ученый, а помогает ему в этом геномный браузер.
Биоинформатикам-разработчикам геномный браузер помогает анализировать сложные случаи, чтобы найти ошибки в работе алгоритмов и понять, как их можно улучшить.
Новый геномный браузер NGB (New Genome Browser) от ЕРАМ работает в вебе, но по скорости и функционалу не уступает десктопным аналогам. Это продукт, которого не хватало на рынке: предыдущие онлайновые инструменты работали медленнее и умели делать меньше, чем десктопные. Сейчас многие клиенты выбирают веб-приложения из соображений безопасности. Онлайн-инструмент позволяет ничего не устанавливать на рабочий компьютер ученого. С ним можно работать из любой точки мира, зайдя на корпоративный портал. Ученому не обязательно всюду возить за собой рабочий компьютер и скачивать на него все необходимые данные, которых может быть очень много.

Геннадий Захаров, бизнес-аналитик:
«Над опенсорсными утилитами я работал частично как заказчик: ставил задачу. Я изучал лучшие решения на рынке, анализировал их преимущества и недостатки, искал, как можно их усовершенствовать. Нам нужно было сделать веб-решения не хуже десктопных аналогов и при этом добавить в них что-то уникальное.
В 3D-просмотрщике молекул это была работа с виртуальной реальностью, а в геномном браузере – улучшенная работа с вариациями. Мутации бывают сложными. Перестройки в раковых клетках иногда затрагивают огромные области. В них появляются лишние хромосомы, куски хромосом и целые хромосомы исчезают или объединяются в случайном порядке. Отдельные куски генома могут копироваться по 10–20 раз. Такие данные, во-первых, сложнее получить из прочтений, а во-вторых, сложнее визуализировать.
Мы разработали визуализатор, который правильно читает информацию о таких протяженных структурных перестройках. Еще мы сделали набор визуализаций, который при контакте хромосом показывает, образовались ли из-за этого контакта гибридные белки. Если протяженная вариация затрагивает несколько белков, мы по клику можем рассчитать и показать, что происходит в результате такой вариации, какие гибридные белки получаются. В других визуализаторах ученым приходилось отслеживать эту информацию вручную, а в NGB – в один клик».
Как изучать биоинформатику
Мы уже говорили, что биоинформатики – гибридные специалисты, которые должны знать и биологию, и информатику. Самообразование играет в этом не последнюю роль. Конечно, в ЕРАМ есть вводный курс в биоинформатику, но рассчитан он на сотрудников, которым эти знания пригодятся на проекте. Занятия проводятся только в Санкт-Петербурге. И всё же, если биоинформатика вам интересна, возможность учиться есть:
1) Вводный курс в генетическую диагностику от компании 23andme.
2) Несколько курсов на Coursera (в том числе пара курсов на русском: введение в биоинформатику и в метагеномику).
3) Курсы на Stepik от института биоинформатики: молекулярная биология и генетика, молекулярная филогенетика, генная инженерия и введение в технологии высокоэффективного секвенирования. Полный список курсов от института можно посмотреть на его официальном сайте.
4) Лекции Павла Певзнера – профессора Калифорнийского университета в Сан-Диего, специалиста в области биоинформатики.
5) Если вы живете в Санкт-Петербурге, можно прийти на гостевые лекции в институт биоинформатики – это бесплатно.
