Перевод поста Пола-Жана Летурно (Paul-Jean Letourneau) "Searching Genomes with Mathematica and HadoopLink".
Код, приведенный в статье, можно скачать здесь.
Примечание: этот пост написан как продолжение поста Большие массивы данных в Mathematica с HadoopLink.
Примечание переводчика: автор данной статьи под термином геном понимает всю совокупность генов некоторого структурного элемента живой материи. Это несколько отличается от стандартных определений, близких по смыслу, в которых подразумевается либо вся совокупность генов конкретного вида (Ridley, M. (2006). Genome. New York, NY: Harper Perennial), либо полный набор генетических инструкций, которые можно найти в клетке (http://www.genome.gov/Glossary/index.cfm?id=90). В данном посте будем пользоваться представлением автора.
В моём предыдущем посте я описал, как писать алгоритмы MapReduce (вики) в Mathematica с помощью пакета HadoopLink. Теперь давайте копнём немного глубже и напишем более серьёзный алгоритм MapReduce.
Я уже писал раньше о некоторых занятных возможностях в сфере геномики в Wolfram|Alpha. Если вам это интересно, вы даже можете осуществлять поиск по человеческому геному определённых последовательностей ДНК. Биологам часто требуется найти расположение фрагмента ДНК, которые они нашли в лаборатории, для определения того, какому животному принадлежит этого фрагмент, или из какой он хромосомы. Давайте используем HadoopLink для создания геномной поисковой системы!
Как и прежде, мы загружаем пакет HadoopLink:
И устанавливаем ссылку на главном узле Hadoop:

Чтобы проиллюстрировать идею, возьмём небольшой по размерам человеческий митохондриальный геном из GenomeData:

Сперва разделим геном на отдельные основания (A, Т, С, G):

Они будут нашими парами ключ-значение (k1, v1). Значения — начальные позиции каждого основания генома:
{k1, v1} = {основание, позиция}
То есть мы только что создали индекс для генома.
Экспортируем этот индекс в Hadoop Distributed File System (HDFS):
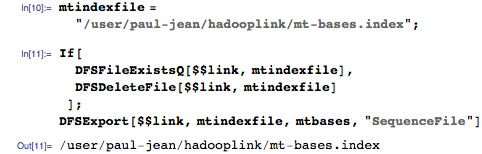
Для запросов мы будем использовать последовательность из 11 оснований, о которых мы знаем, что они содержатся в геноме:

Мы используем последовательность с большим количеством повторений в ней для того, чтобы усложнить задачу нашим алгоритмам.
Наш геномный поисковик на этот запрос должен вернуть позицию 515:
Теперь нам нужны mapper и reducer.
Как было сказано в первой части — mapper принимает пару ключ-значение (шаг 1), и выдаёт другую пару (шаг 2):

Mapper получает основание из индекса генома в качестве входного значения и выдаёт пары ключ-значение для каждого расположения в запросе, где встречаются индексы оснований (скоро вы узнаете почему):
(1) Ввод: {k1, v1} = {индекс основания, позиция в геноме}

Ключ на выходе — положение в геноме, значение на выходе — положение в запросе:
(2) Вывод: {k2, v2} = {позиция в геноме, позиция в запросе}
Какая разница между позицией в геноме и позицией в запросе? Положение в запросе есть положение основания в запросе, в то время как положение в геноме есть положение в целом геноме.
К примеру, скажем, mapper получает пару ключ-значение с основанием A на позиции 517:
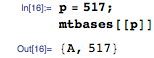
Позиции в запросе для основания А в запрашиваемой последовательности GCACACACACA есть 3, 5, 7, 9, и 11:
Вот эта последовательность с подсвеченными позициями:

Mapper имеет только одну пару ключ-значение с одной базой индексов вместе с запрашиваемой последовательностью. У него не имеется остальной части генома для сравнения, так что ему требуется найти все возможные способы выстраивания запроса с основанием A на позиции 517:

Здесь цвета соответствуют каждой A в запросе (по горизонтали) с их получающимися положениями в геноме (по вертикали). Возьмем, к примеру A в третьем основании в запросе (показано зеленым). Если его встроить с A на позиции 517, то запрашиваемая последовательность будет начинаться в геноме с позиции 515 (517 — 3 + 1 = 515) (также показано зеленым).
Точно так же, выделенное красным основание (пятое положение в запросе) заставит запрашиваемую последовательность выстраиваться, начиная с позиции 513 в геноме (также показано в красным). И то же самое для седьмой позиции в запросе с 511 позицией в геноме, (фиолетовый) девятой позиции в запросе с 509 позицией в геноме (оранжевый), одиннадцатой позиции в запросе с 507 позицией в геноме (коричневый).
Лишь одна из этих расстановок является правильной. В данном случае — для третьей позиции в запросе (зеленый цвет), которая позволяет запрашиваемой последовательности встроиться в геном. Но mapper этого не знает, и потому рассматривает все возможные совпадения.
Так так reducer собирает ключи, он соберёт и все основания, которые соответствуют одним и тем же позициям в геноме:
Ввод: {k2, {v2 …}} = {позиция в геноме, {позиции в запросе…}}
Теперь для данного положения в геноме reducer будет искать те случаи, когда значения формируют полную последовательность из позиций запросов:

Если reducer находит полное соответствие, он выдаёт положение генома:
Вывод: {k3, v3} = {запрашиваемая последовательность, положение в геноме}
Давайте теперь запросим у нашего геномного поисковика последовательность GCACACACACA:
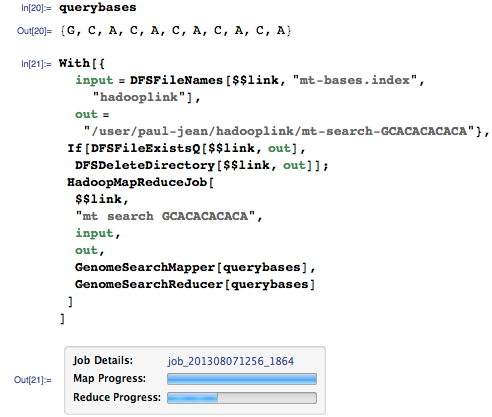
(См. часть 1, где приводилось описание функции HadoopMapReduceJob).
И импортируем соответствия в геноме из HDFS:
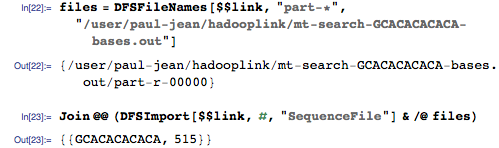
Соответствующее положение в геноме — 515, и это правильный ответ! Наша геномная поисковая система работает!
Давайте теперь осуществим поиск с запросом, которому должны соответствовать два разных положения в геноме:

Этому запросу должны соответствовать позиции 10 и 2277:
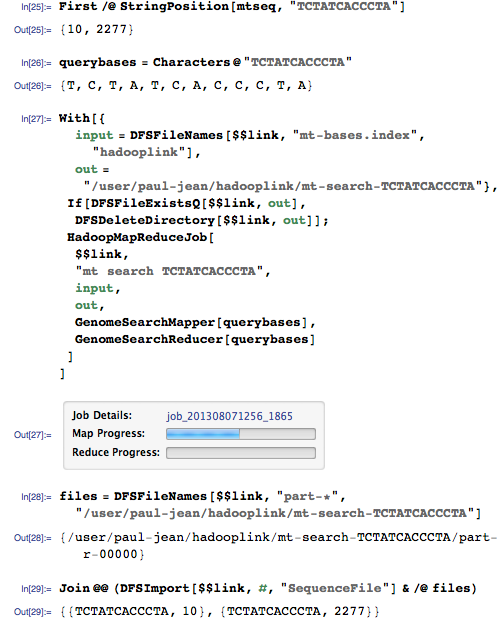
Да, он нашел оба совпадения!
Теперь давайте отмасштабируем всё это для всего человеческого генома. Первым шагом является создание индексации, на этот раз для всего генома, а не только митохондриального. Чтобы это сделать, я скачал весь человеческий геном в виде текстовых файлов с государственного сервера и импортировал их в HDFS:

Получается по одному текстовому файлу на хромосому, содержащие необработанные последовательности хромосом:
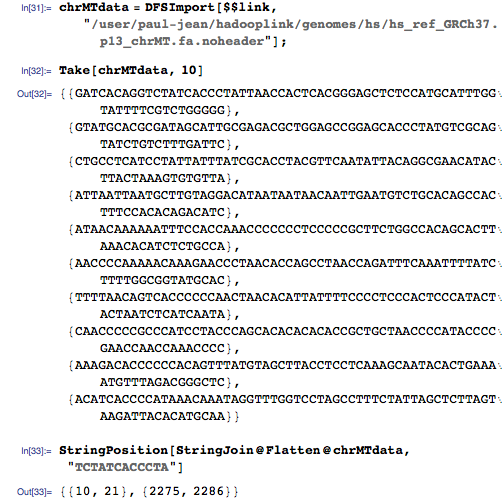
Затем я применил MapReduce для создания пар ключ-значение в индексе на HDFS, который выглядит так:
[hs_ref_GRCh37.p13_alts.fa, 121] G
[hs_ref_GRCh37.p13_alts.fa, 122] A
[hs_ref_GRCh37.p13_alts.fa, 123] A
[hs_ref_GRCh37.p13_alts.fa, 124] T
[hs_ref_GRCh37.p13_alts.fa, 125] T
[hs_ref_GRCh37.p13_alts.fa, 126] C
[hs_ref_GRCh37.p13_alts.fa, 127] A
[hs_ref_GRCh37.p13_alts.fa, 128] G
[hs_ref_GRCh37.p13_alts.fa, 129] C
[hs_ref_GRCh37.p13_alts.fa, 130] T
Одно небольшое отличие от предыдущего примера в том, что ключ сейчас представляет собой {хромосома, позиция в геноме}, а значение — основание на этой позиции. Так что теперь хромосома будет в ключе. Я немного изменил mapper, чтобы он мог работать с новым представлением ключа:

Reducer остался без изменений.
Давайте запустим поиск для той же последовательности:
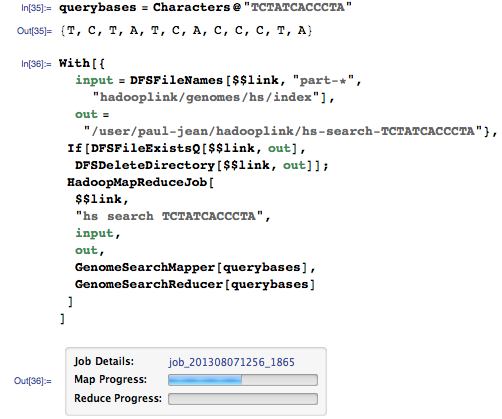
На этот раз мы получаем совпадения для всего генома:
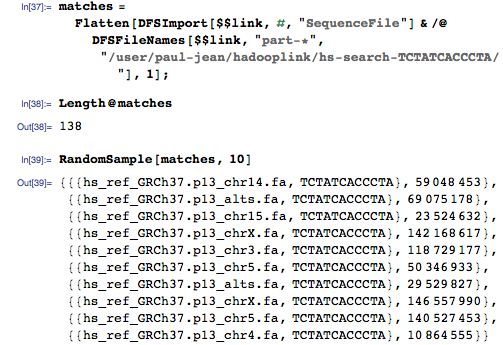
И мы можем ещё улучшить алгоритм. Как насчёт поиска неточных соответствий вместо точных? Слегка поменяем reducer, указав то, насколько большая часть запроса должна совпадать:

Это не самый эффективный способ поиска в геноме, но он показывает, как легко писать прототипы алгоритмов на основе MapReduce и запускать их в Mathematica. Если вам интересны подробности, рекомендую посмотреть мои недавние сообщения. А на интересующие вопросы вам с радостью ответят в HadoopLink GitHub Repo!
